Частота заболевания
СМА – одно из самых часто встречеющихся заболеваний из орфанных (редких), болеет один новорожденный на 6000-10000 .
Причина СМА
СМА — наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
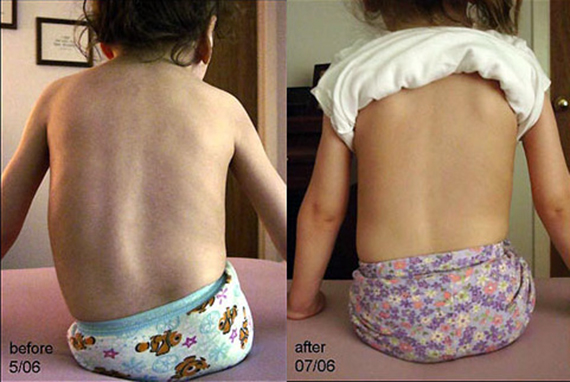
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.

От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III , болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА
IV
, этот тип называется ещё «взрослой СМА»,
поскольку болезнь проявляется обычно в возрасте после 35 лет.
Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.
Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук. Обычно проблем с глотательной и дыхательной функцией у больных нет.
Большинство из больных IV типом СМА могут ходить, и лишь некоторым приходится прибегать к инвалидным коляскам.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
На сегодняшний момент радикального лечения от СМА не существует.
Международной корпорацией «Биоген» был разработан препарат «Спинраза», который значительно улучшил состояние больных, к которым применялся во время тестирования. В настоящее время препарат одобрен к применению в США, в Европе ориентировочная стоимость годового курса, по подсчётам компании, будет составлять около 270 тысяч евро, в России препарат не сертифицирован. Лечение пожизненное.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.

Фонд работает по всей территории России. Работа фонда имеет два основных направления — оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования — например, СМА 300 .
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно. В этом отношении СМА – такая же «отложенная» болезнь как, например, миодистрофия Дюшенна или синдром Ретта, когда ребёнок, некоторое время развивавшийся в соответствии с нормой, теряет приобретённые ранее навыки и становится инвалидом.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
Сколько больных СМА в России?

Препарат «Спинраза», значительно улучшающий состояние больных. Фото с сайта healthbeat.spectrumhealth.org
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Кто в России помогает людям со СМА и их семьям
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Известные люди со СМА

Итальянка Симона Спиноглио
родилась с наследственным заболеванием – спинальной мышечной атрофией 2 типа. Она с самого рождения не может ходить и передвигается только с помощью электрической коляски. Но ее жизнь полна и насыщенна; ничто не может помешать ее стремлению жить.
Симона работает на «горячей линии» итальянской Ассоциации «Семьи СМА» (Famiglies of SMA) и помогает детям и взрослым со СМА и другими нервно-мышечными заболеваниями.
Также Симона записала несколько популярных в итальянском сообществе СМА песен — о свободе делать то, что ты хочешь, несмотря на болезнь.
Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
В 2008 году собрала собственную музыкальную группу (распалась в 2010). В 2013 году приняла участие в конкурсе «Фактор А» на телеканале «Россия». Заняла второе место и получила персональную премию Аллы Пугачёвой «Золотая звезда Аллы». В 2017 году из-за недопуска России в конкурсную программу не смогла принять участие в конкурсе «Евровидение». Передвигается на коляске.
Программист из Владимира Валерий Спиридонов . Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Сегодня Валерий — член городской общественной палаты Владимира, эксперт по вопросам доступной среды, а также создатель собственного сообщества «Desire for life», рассказывающего о создании доступной среды и перспективных медицинских проектах. Валерий – участник многих телепрограмм на российском и зарубежном ТВ.
Спинальная мышечная атрофия (СМА), или амиотрофия, — это заболевание наследственного характера, которое сопровождается острыми нарушениями в активности нейронов мозга спинного и головного. Процессы затрагивают мотонейроны. Впервые заболевание описали в соответствии с медицинской картиной в XIX веке. Оно относится к группе генетических нарушений, обусловленных мутациями.
Специфичность мышечной атрофии заключается в том, что только один вид спинально патологии – первый – развивается у новорожденного в течение 1-2 месяца жизни. Остальные формы болезни дают о себе знать только во взрослом возрасте. Сложную форму спинальной атрофии и методы ее лечения изучают такие дисциплины, как генетика, неврология и педиатрия.
Существуют разные данные о том, как часто встречается спинально-мышечная атрофия у новорожденных детей. Плотность случаев напрямую связана с населенностью того или иного места на планете. Из-за того, что патологию часто обнаруживают только во взрослом возрасте, количество случаев после 20 лет больше, чем во младенчестве. Примерно 1 человек из 20 000 страдает одной из форм нарушения.
Факт! Среди младенцев тяжелые формы спинальной болезни встречаются в среднем 5-7 раз на 100 000 человек.
Наследственный фактор же проявляется далеко не у всех. Так, родители могут оказаться носителями мутировавшего гена. Но проявится он только у ребенка с вероятностью в 50-70%. Считается, что распространенность СМА среди носителей – 1 на 80 семей, или на 160 человек разного пола.
СМА – это одна из самых распространенных форм дегенеративных процессов наследственного характера у детей. Она занимает 2 место после муковисцидоза и считается причиной №1 среди наследственных болезней, ведущих к смерти ребенка до достижения им 15-18 лет.

Летальный исход возникает на фоне дыхательной недостаточности. Чем раньше спинальная патология проявляет себя, тем хуже будет прогноз. В среднем, дети с мышечно-спинальной атрофией доживают до 10-11 лет. При этом состояние интеллекта не оказывает влияния на прогресс спинальной амиотрофии.
Нарушение встречается чаще у мальчиков, чем у девочек, и протекает у них гораздо сложнее. На 1 пациента женского пола приходится 2 пациента – мужского. Но с 8 лет учащение среди девочек увеличивается.
Генетические факторы болезни
Спинальная мышечная атрофия появляется при наследовании рецессивного генома 5 хромосомы. Если оба человека, которые родили младенца, являются носителями СМА, то с вероятностью не менее 25% передают ген ребенку. В результате нарушается синтез белковых структур, разрушение моторных нейронов спинного мозга происходит в несколько раз быстрее, чем восстановление.
В период эмбрионального развития нервная система ребенка производит только половину от необходимого объема моторных нейронов. Со временем при СМА этот процесс сильно замедляется. После рождения из-за недостатка структур развивается спинальная атрофия.

Особенности функционирования нейронов
Активный мозг постоянно посылает импульсы в спинной мозг, а проводниками служат нервные клетки. Они доставляют сигналы в мышцы, в результате чего запускается их движение. Если этот процесс нарушен, то движение становится невозможным.
При спинально-мышечной атрофии двигательные нейроны ног, входящие в состав спинного мозга, работают некорректно. Они отвечают за сигналы, с помощью которых мозг поддерживает такие функции, как ползанье, поддержка шеи, сжимание и движение руками, ногами, а также дыхание и глотательный рефлекс.
Важно! При получении дефектных копий гена SMN1 от родителей, нервная система ребенка перестает производить белок, который контролирует процесс синтеза и обмена нейронов.

В результате мышцы, не получающие постоянных сигналов, начинают атрофироваться.
Классификация типов атрофии
Существует 4 распространенных группы спинально-мышечной атрофии у детей и взрослых:
- Младенческая форма. Самый сложный тип мышечно-спинальной атрофии, называемый также патологией Верднига-Гоффмана. Течение патологии при этой форме осложняется быстрым развитием тяжелых симптомов: появляются трудности с глотанием, сосанием и дыханием. Младенцы с СМА1 не могут нормально держать голову, сидеть.
- Промежуточная форма. СМА2, или болезнь Дубовица, несколько отличается степенью тяжести. При этой форме патологии ребенок может сохранять сидячее положение и даже есть, так как глотательные функции частично не нарушены. Но ходить он не сможет. Прогноз напрямую связан со степенью повреждения респираторных мышц, отвечающих за активность легких.
- Юношеская форма. СМА3, или болезнь Кюгельберга-Веландер, переносится подростками проще, чем первые типы спинально-мышечной атрофии. Ребенок может стоять, однако будет страдать от сильной слабости. Высок риск инвалидности – потребность в коляске сохраняется у большинства.
- Взрослый тип. СМА4 встречается в основном после 35 лет. Продолжительность жизни при заболевании не изменяется, но у пациента появляется выраженная слабость мускулатуры, снижение сухожильных рефлексов. По мере прогрессирования, требуется инвалидное кресло.

Заподозрить спинально-мышечную патологию сразу же после рождения очень сложно. Но обнаружение на первых этапах способно облегчить страдания пациентов, поэтому нужно знать о распространенных симптомах спинально-мышечной атрофии.
Симптомы разных форм болезни
Существует общий набор признаков СМА, по которым можно заподозрить патологию, если других проблем не обнаруживается, либо диагноз вызывает сомнение. Группу симптомов сводят к проявлению вялого периферического паралича:
- выраженная мышечная слабость или атрофия разных мышечных групп;
- сначала в процесс вовлекаются конечности – симметрично, ноги, а затем руки, постепенно втягивается и туловища;
- отсутствуют расстройства чувствительности и тазовые нарушения;
- наиболее выраженные проблемы затрагивают проксимальные или дистальные мышечные группы.

У пациентов появляются подергивания и фибрилляции – мерцательные аритмии.
Признаки СМА1
Заболевание Верднига-Гоффмана бывает 3 видов:
- Врожденная форма. Начинается в течение 1-6 месяца жизни, обладает тяжелыми симптомами. Обнаружить признаки можно во внутриутробном развитии – эмбрион будет мало двигаться. Гипотония наблюдается сразу же после рождения ребенка. Такие младенцы не держат голову, не могут сидеть. Постоянно находятся в позе лягушки с раздвинутыми конечностями. Сначала симптомы появляются в ногах, затем в руках, после этого страдает дыхательная мускулатура. Психическое развитие у таких детей медленное, они редко доживают до 2 лет.
- Ранняя спинальная мышечная атрофия. Первые признаки начинают беспокоить пациента до 1,5 лет, чаще всего – после какой-либо инфекции. Даже если раньше ребенок мог стоять и сидеть, теперь он утрачивает эти функции. Развиваются парезы, а затем поражаются дыхательные мышцы. Ребенок умирает, как правило, в результате затяжной пневмонии или дыхательной недостаточности в возрасте 3-5 лет.
- Поздняя форма. Патология встречается после 1,5 лет, двигательные возможности сохраняются у ребенка до 10 лет. Медленный прогресс симптомов приводит к дыхательной недостаточности и смерти в возрасте до 18 лет.

СМА1 – наиболее тяжелая форма патологии, готовиться всегда приходится к худшему исходу.
Признаки болезни Кугельберга-Веландера
Возникает в возрасте от 2 до 15 лет. Сначала в процесс вовлекаются нижние конечности, затем пояс таза, на последних стадиях страдает плечевой пояс и дыхательная система. Примерно у 25% пациентов появляется синдром псевдогипертрофии мышц, из-за чего патологию путают с мышечной болезнью Беккера.
Спинальная мышечная атрофия Кугельберга-Веландера не сопровождается костными деформациями, и пациенты способны сами себя обслуживать в течение долгих лет.

Амиотрофия Кеннеди
Эта патология входит во взрослую группу, болеют представители мужского пола после 30 лет. Женщины от патологии не страдают. Течение – умеренное, сначала поражаются ножные мышцы, последующие 10-20 лет пациент сохраняет привычный ритм жизни. Только после этого начинают страдать мускулы рук и головы. У многих пациентов со временем наступают эндокринные изменения: атрофия яичек, отсутствие либидо, сахарный диабет.

Дистальная СМА
Эта форма спинально-мышечной атрофии также развивается у взрослых пациентов после 20 лет. Ее второе название – СМА Дюшенна-Арана. Риск развития патологии сохраняется вплоть до 50 лет. Атрофия начинается в руках, вызывает синдром «когтистой лапы», потом переходит на крупные мышцы. Со временем появляются парезы мышц нижних конечностей, а туловище страдает редко. Прогноз у этой формы благоприятный, если не присоединяется торсионная дистония или болезнь Паркинсона.

СМА Вюльпиана
Скапуло-перонеальная форма спинально-мышечной атрофии, сопровождаемая симптомом «крылатых» лопаток. Появляется в среднем в 20-40 лет, позже встречается реже. Поражается плечевой пояс, а через некоторое время – руки и нижние конечности. При этой форме спинальной болезни двигательные функции у пациента сохраняются на 30-40 лет.

Способы диагностики патологии
Распознать спинально-мышечную атрофию со 100% гарантией можно только с помощью анализа ДНК на молекулярно-генетические факторы. С его помощью можно найти дефективный ген в 5 хромосоме.
Также применяют биохимический анализ для выявления состояния белка. Электрофизиологическое исследование мозга необходимо для определения активности импульсов и нервных стволов. МРТ и КТ назначают редко, так как эти методы не имеют высокой эффективности.

Методы лечения
Эффективных способов лечения спинально-мышечной атрофии не существует. Однако легкие стадии поддаются коррекции. С помощью физиотерапии, массажа и медикаментов можно поддерживать комфортное состояние ребенка. Во взрослом возрасте терапия более эффективна, так как эти формы атрофии не настолько тяжело переносятся.
Медикаменты
Для коррекции работы мышечных волокон и нервных импульсов используют препараты, улучшающие кровообращение и замедляющие разрушение нейронов:
- Антихолинэстераза. Средства подавляют активность фермента, расщепляющего ацетилхолин: «Прозерин», «Оксазил», «Сангвиритрин».
- Витамины и БАД. Используют антиоксиданты, карнитин, витамины группы B для поддержания обмена веществ и тонуса.
- Ноотропы. Улучшают работу нервной системы: «Ноотропил», «Кавитон», «Семакс».
- Средства для активации обмена веществ. В эту группу входят различные продукты: никотиновая кислота, «Актовегин», «Калия оротат».

Также важно поддерживать правильное питание ребенка, не допускать злоупотребления жирами и рафинированными продуктами.
Физиотерапия
Физиотерапевтические процедуры при спинально-мышечной атрофии улучшают тонус, кровообращение, обмен веществ, помогают снизить болезненные ощущения. Назначают: УВЧ, электрофорез, мануальные техники, дыхательные аппараты для стимуляции легких.
Внимательный контроль дыхания
Так как спинальная мышечная атрофия часто связана с такими нарушениями, как дыхание, необходимо строго следить за функционированием этой системы у ребенка:
- назначают физиотерапию грудной клетки;
- очищают дыхательные пути от образовывающейся слизи;
- назначают обезболивающие;
- принимают препараты, снижающие выработку секрета;
- используют методы неинвазивной вентиляции легких, которые повышают комфорт пациента и предотвращают гиповентиляцию ночью;
- применяют инвазивные методы – искусственную вентиляцию с помощью введенной трубки.

Последний метод используется в тяжелых случаях, когда дыхательный рефлекс становится невозможен.
Питание ребенка
Если спинальная мышечная атрофия развилась до такой степени, что пациент больше не может самостоятельно глотать, ему требуется сторонняя помощь. Нужно корректировать мышечную слабость.
Подробно о том, как кормить маленького пациента с нарушением глотательных функций, рассказывает доктор, ведущий мышечную атрофию. Иногда для реализации этих целей требуется профессиональная медицинская помощь.
Важно! При лечении больных с СМА не требуется соблюдение жесткого рациона или введения/ограничения каких-либо продуктов с содержанием определенных веществ, витаминов и минералов.
У детей с СМА может быть нарушен процесс пищеварения, из-за чего дети начинают мучиться от запоров. Иногда развивается рефлюкс-болезнь.
Прогноз и возможные последствия
Если спинальная мышечная атрофия обнаружена у пациента во взрослом возрасте, то прогноз более благоприятный. Патология СМА1 редко оставляет надежды – большая часть детей не доживает до 2 лет, остальные погибают в возрасте до 5 лет.
Смерть наступает из-за дыхательной недостаточности, реже – из-за острой, не проходящей, пневмонии. В настоящее время способов профилактики заболевания не существует.
Взрослые люди с диагнозом СМА должны отказаться от вредных привычек, экстремальных видов спорта и ненормированного режима отдыха/труда. Это значительно замедлит прогресс спинально-мышечной болезни.
СА представляют собой гетерогенную группу наследственных заболеваний периферической нервной системы, которые характеризуются выраженным клиническим полиморфизмом.
Спинальная мышечная атрофия (или СА) - разнородная группа наследственных заболеваний, протекающих с поражением и потерей моторных нейронов передних рогов спинного мозга.
Амиотрофия - это нарушение трофики мышц, сопровождающееся истончением мышечных волокон и уменьшением их сократительной способности, обусловленное поражением нервной системы: мотонейронов (на различных уровнях ЦНС - нейроны двигательной коры, ядер ствола мозга, передних рогов спинного мозга) или периферических нервных волокон. Болезнь считается наследственной в результате генных мутаций, хотя если мы посмотрим истории болезни, то у многих больных семейный анамнез не выявляется.
Различают наследственные и симптоматические амиотрофии. Нейрогенные наследственные амиотрофии делятся на две большие группы - спинальные и невральные амиотрофии. В большинстве случаев более тяжелыми являются спинальные формы. К ним относятся: спинальная амиотрофия (болезнь Верднига - Гоффманна), псевдомиопатическая прогрессирующая спинальная амиотрофия Кугельберга - Веландер, редкие формы спинальных амиотрофий и недифференцированные формы. Невральные амиотрофии: болезнь Шарко - Мари - Тута, гипертрофическая невропатия Дежерина - Сотта, синдром Русси - Леви, атактическая полинейропатия или болезнь Рефсума, а также недифференцированные формы.
СА делятся также на взрослые и детские. К проксимальным СА детского возраста относятся: острая злокачественная инфантильная СА Верднига - Гоффманна (спинальная амиотрофия 1-го типа), хроническая инфантильная СА (спинальная амиотрофия 2-го типа), ювенильная СА (болезнь Кугельберга - Веландер), редкие формы СА в детском возрасте: инфантильная нейрональная дегенерация, врожденная форма болезни Пелицеуса - Мерцбахера, врожденная цервикальная СА, атипичный вариант GM-ганглиозидоза, детский прогрессирующий бульбарный паралич (синдром Фацио - Лонде), понтобульбарный паралич с глухотой (синдром Виалетто - Ван Лэре).
СА взрослых: бульбоспинальная амиотрофия Кеннеди, дистальная СА, сегментарная СА, мономиелическая СА, скапуло-перонеальная СА Старка - Кайзера, лицелопаточно-плечевая СА Феничела, окулофарингеальная СА. Существуют также недифференцированные формы СА с быстропрогрессирующим, медленнопрогрессирующим и непрогрессирующим течением.
Согласно рекомендации Европейского консорциума по изучению нервно-мышечных заболеваний, клиническими критериями спинальной мышечной амиотрофии являются: [1 ] симметричная мышечная гипотония и гипотрофия, [2 ] фасцикуляции различных мышечных групп, [3 ] гипо- или арефлексия мышц конечностей, [4 ] отсутствие чувствительных, мозжечковых и интеллектуальных расстройств.
Обратите внимание ! Патогномоничных изменений при спинальной мышечной амиотрофии нет. Однако важным является определение активности креатинкиназы сыворотки крови: считается, что превышение ее нормы более чем в 10 раз характерно для миодистрофии и противоречит диагнозу спинальной мышечной амиотрофии.
читайте также пост: Креатинкиназа (справочник невролога) (на сайт)
При электронейромиографии (ЭНМГ) выявляются симптомы поражения периферических моторных нейронов: спонтанная мышечная активность, увеличение длительности и амплитуды потенциалов действия двигательных единиц при нормальной скорости проведения импульсов по афферентным и эфферентным волокнам периферических нервов. При гистологическом исследовании биоптатов мышц обнаруживаются признаки денервационной мышечной атрофии.
Классическая проксимальная СА взрослых начинается на 3-м десятилетии жизни и наследуется по аутосомно-рецессивному типу. СА обычно дебютирует в 40 - 50 лет , однако встречаются случаи с началом в подростковом возрасте. Распределение мышечной слабости при аутосомно-доминантном типе в ряде случаев значительно шире, чем при аутосомно-рецессивном типе. Проксимальные мышцы также поражаются тяжелее, чем дистальные. Симптомы прогрессируют медленно , двигательные функции и способность ходить у подавляющего числа больных сохраняются и в зрелом и даже в старческом возрасте. Слабость бульбарных мышц не характерна . Глазодвигательные мышцы не поражаются . Сухожильные рефлексы угнетены или отсутствуют. Контрактуры суставов редки . Уровень КФК нормальный или незначительно повышен. Будут рассмотрены следующие формы СА взрослых:
1. бульбоспинальная амиотрофия Кенеди;
2. дистальная СА;
3. сегментарная СА;
4. мономиелическая СА;
5. скапулоперониальная СА Старка-Кайзера;
6. лицелопаточноплечевая СА Феничела;
7. окулофарингеальная спинальная амиотрофия.
Бульбоспинальная амиотрофия Кеннеди. Редкая Х-сцепленная форма спинальной амиоторфии; дебютирует на 4-й декаде жизни, хотя изредка отмечаются случаи первых проявлений в 12 - 15 лет. Кен картирован на длинном плече Х-хромосомы в сегменте Хq21-22. Мутация затрагивает ген андрогенного рецептора и представляет собой экспансию нуклеотидного триплета (цитозин - аденин - гуанин). Ядро клинической картины болезни составляют слабость, атрофии и фасцикуляции в проксимальных мышечных группах конечностей, сухожильная арефлексия, слабость мимических мышц, атрофии и фасцикуляции в языке, периоральные фасцикуляции, дизартрия и дисфагия (последняя не является прогностически неблагоприятным признаком) постуральный тремор и крампи. Изредка развивается аксональная невропатия. Бульбарные нарушения возникают, как правило спустя 10 лет после начала заболевания. Характерны эндокринные нарушения: гинекомастия (!), тестикулярная атрофия, снижение потенции и либидо, сахарный диабет. Треть больных страдает бесплодием, обусловленным азооспермией. Проявления феминизации и гипогонадизма, вероятно связаны с нечувствительностью дефектных андрогенных рецепторов к мужским половым гормонам (их уровень у больных остается нормальным). Прогноз болезни в целом благоприятен. Сохраняется ходьба и возможность самообслуживания. Продолжительность жизни не укорочена. Однако существует повышенный риск злокачественных опухолей вследствие гормонального дисбаланса (рак молочной железы), что требует онкологической настороженности. Заболевание необходимо отличать от БАС. В настоящее время возможно проведение прямой ДНК-диагностики болезни, установление гетерозиготного носительства и осуществление дородовой диагностики ( о бульбоспинальной амиотрофии Кеннеди на сайт).
Дистальна СА. Аутос.-рецес. форма может начинаться в раннем детском возрасте, тогда как аутос.-домин. форма - в 23 - 25 лет. При обоих типах наследования могут быть представлены и тяжелые клинические формы, и формы средней тяжести. Заболевание начинается со слабости и атрофии передней группы мышц голеней, которым сопутствуют деформации стоп. Сухожильные рефлексы могут быть сохранены. Клиническая картина может напоминать НМСН Ι типа, однако при СА чувствительность не нарушается. При тяжелых аутос.-рецес. формах мышечная слабость постепенно распространяется на проксимальные мышцы ног, а иногда и рук. Степень слабости в руках варьирует между разными семьями, но практически одинакова у представителей одной семьи. Примерно у 25% больных обнаруживают сколиоз. В отдельных семьях у пораженных может выявляться псевдогипертрофия или атрофия икроножных мышц. Данные ЭНМГ позволяют отличить заболевание от периферической невропатии: скорость проведения по двигательным аксонам нормальная, несмотря на признаки тотальной денервации мелких мышц стопы. Вызванные сенсорные потенциалы также нормальны. Уровень КФК нормальный, иногда умеренно повышен.
Сегментарная СА: поражаются только руки или только стопы; заболевание характеризуется генетической гетерогенностью: аутос.-дом. наследование типично для формы с дебютом у взрослых; аутос.-рецес. - для формы, начинающейся у подростков, в основном мальчиков. Атрофии кистей, как правило, асимметричные, прогрессируют в течение 2 - 4 лет и иногда поражают и предплечья. Характерны фасцикуляции и крампи. Обычно нарастание артофий со временем прекращает, но в некоторых случаях вовлекаются мышцы ног.
Мономелическая СА: при этой редкой форме поражаются мышцы руки или ноги. Большинство случаев зарегистрировано в Японии и Индии. Мономелическая СА обычно встречается в виде спорадических случаев с превалированием мужчин 10: 1, что позволяет предположить Х-сцепленный рецессивный тип наследования. Возраст дебюта варьирует от 10 до 25 лет. Слабость и атрофия мышц нарастают незаметно. Рука поражается чаще, чем нога. Слабость может быть распределена только проксимально, только дистально или захватывать всю конечность. Атрофия вначале односторонняя и наблюдается в мышцах, иннервируемых С7, С8 и Th1 спинальными сегментами. Двусторонняя мышечная слабость обыкновенно развивается в течение 2 лет. Часто наблюдается односторонний или двусторонний постуральный тремор кистей. Фасцикуляции в проксимальных мышечных группах предшествуют появлению слабости и атрофии. Прогрессирование болезни медленное и через 5 лет, как правило, наступает стабилизация. Однако спустя 15 лет в патологический процесс может быть вовлечена другая конечность. Должны быть исключены другие причины моноплегии.
Скапулоперонеальная СА Старка-Кайзера. Эта редкая форма СА является генертически гетерогенной. Случаи, наследуемые аутосомно-доминантно, дебютируют на 3-4-й декаде жизни и отличаются относительно доброкачественным течением, тогда как случаи с аутосомно-рецессивным наследованием - дебютируют в 3 - 5 лет. Предполагается сцепление с локусом 12q24. У некоторых больных определяется мутация в SMN гене 5-й хромосомы, что ставит под сомнение нозологическую самостоятельность ряда случаев скапулоперонеальной СА и свидетельствует о своеобразном варианте экспрессии гена проксимальной СА. Слабость и атрофии мышц превалируют в плечелопаточной группе мышц и разгибателях стопы. Возможно медленное распространение атрофий на проксимальные отделы ног и мускулатуру тазового пояса. Дифференциальную диагностику проводят со скапулоперонеальной миодистрофией.
Лицелопаточноплечевая СА Феничела. Редкая аутосомно-рецессивная форма СА, начинающейся на 2-м десятилетии жизни. Ген пока не картиован. Заболевание имитирует лицелопаточно-плечевую миодистрофию Ландузи-Дежерина, однако при нем сухожильные рефлексы обычно вызываются, а сила мышц снижается незначительно. На ЭМГ регистрируется нейронально-аксональный тип поражения. Активность КФК нормальная. Ряд исследователей оспаривает нозологическую самостоятельность данной формы и рассматривает ее в рамках болезни Ландузи-Дежерина.
Окулофарингеальная СА. Предполагается аутосомно-доминантный тип наследования. Заболевание обычно начинается на 4-м десятилетии жизни с наружной офтальмоплегии, дисфагии и дизартрии. В некоторых случаях присоединяется слабость в дистальных отделах конечностей и мышцах спины. Течение медленное, доброкачественное. Иногда болезнь рассматривается в рамках митохондриальных миопатий.
источник : использованы материалы руководства для врачей "Болезни нервной системы" под ред. Н.Н. Яхно, Д.Р. Штульмана, изд. 2-е, том 1; Москва, "Медицина", 2001 (а также указанных ниже статей).
Спинальная мышечная атрофия является основной генетической причиной смерти в детском возрасте. Давайте разберемся в причинах и способах изменения жизни ребенка, а также узнаем, какие методы лечения доступны на сегодняшний день.
Что такое спинальная мышечная атрофия
Спинальная мышечная атрофия (SMA: Spinal Muscular Atrophy) – это нервно-мышечное заболевание аутосомно-рецессивного типа, характеризуется гибелью двигательных нейронов, расположенных в переднем роге серого вещества спинного мозга и в нижней части ствола головного мозга.
Моторные нейроны – это клетки, из которых образуются нервы, предназначенные для управления скелетными и поперечно-полосатыми мышцами глотки и гортани: когда они вырождаются, целые группы волокон подвергаются атрофии и, соответственно, результатом является мышечная слабость .
Работы глазных мышц, хотя и управляется моторными нейронами энцефального ствола мозга, не нарушается при этой болезни.
Частота спинальной мышечной атрофии колеблется от 1:6000 до 1:10000, и подвержены ей все этнические группы; является редким заболеванием, является одним из самых распространенных нервно-мышечных заболеваний, точнее вторым после дистрофии Дюшенна.
Причина спинально мышечной атрофии
Причина спинально мышечной атрофии была обнаружена в середине 90-х годов, спустя сто лет после первого описания болезни. В 95% случаев речь идёт о делеции в гене SMN1, локализованном на длинном плече хромосомы 5 (делеция – это потеря последовательности ДНК).
Поскольку спинально мышечная атрофия наследуется по аутосомно-рецессивному типу, для развития болезни человек должен получить обе копии плохого SMN1 – от матери и от отца. Таких родителей называют гетерозиготными или носителями, и они не имеют симптомов заболевания. Носители встречаются с частотой 1:50.
Ген SMN1 кодирует белок SMN, который используется в цитоплазме и ядре всех клеток и имеет решающее значение для формирования snRNP, малых ядерных рибонуклеопротеидов, компонентов сплайсинг машин.
Почему же вездесущий белок SMN является критическим фактором для выживания и надлежащего функционирования моторных нейронов?
Другие гипотезы были сформулированы для объяснения антиапоптической роли SMN:
- потребность в этом белке выше у моторных нейронов, чем в других тканях.
- по мнению других авторов, это можно объяснить тем, что белок SMN участвует в транспортировке вдоль аксонов РНК-связывающих белков.
Несмотря на все предположения, в настоящее время ещё неясно, какая из многих функций протеина SMN связана с развитием спинально мышечной атрофии .
4 типа спинально мышечной атрофии
Спинально мышечную атрофию классифицируют на четыре типа, в соответствии :
- с возрастом появления симптомов
- с максимальной двигательной активностью , на которую способен больной
У 25% лиц избегают точной классификации. Кроме того, у людей, страдающих от одного типа заболевания, симптомы могут существенно различаться.
1 тип – болезнь Верднига-Гоффмана
Это самая тяжелая форма спинально мышечной атрофии, составляет 50% от всех случаев.
Главные её особенности:
- проявляется до 6-го месяца жизни
- ребенок имеет плохую и дряблую мышечную массу : он мало движется, потому что не может противостоять силе тяжести, не в состоянии держать голову в вертикальном положении и сидеть без поддержки
- кости хрупкие и подвержены переломам , кроме того, в позвоночнике развивается сколиоз . Проблемы с костями у пациента со спинально мышечной атрофией не удивляют, так как именно физическая активность способствует минерализации костей
- рефлекс сосания и глотания слабый , поэтому такого ребёнка трудно кормить
- грудная клетка ребенка меньше нормы из-за слабости дыхательных мышц. Кашлевый рефлекс слабый, что нарушает процесс избавления от выделений (слизи и твердые частицы, включая микробов)
У детей, страдающих от спинально мышечной атрофии 1 типа, часто развивается пневмония , так как они не в состоянии избавиться от каких-либо патогенных микроорганизмов с кашлем, а также из-за потери контроля глотательных мышц, которые не могут предотвратить попадание слюны и кусочков пищи в легкие. Повторяющие пневмонии ведут, к сожалению, к дыхательной недостаточности.
Для тех, кто страдает от этой формой патологии прогноз неблагоприятный: смерть наступает в течение 2 лет , даже самое хорошее лечение продлевает жизнь только до 5 лет.
2 тип – болезнь Дубовица
Промежуточная форма спинальной мышечной атрофии.
Давайте посмотрим характеристики:
- проявляется между 6 и 18 месяцами
- ребенок показывает задержку в развитии моторики : не в состоянии сидеть, ему нужна поддержка, чтобы стоять, и никогда не научится ходить. Может иметь легкий тремор рук
- при этом типе также отмечается склонность к развитию сколиоза и хрупкости костей
- у некоторых маленьких пациентов дисфагия становится препятствием для поглощения достаточного для развития количества калорий
- кашлевый рефлекс может ослабнуть, облегчая возникновение респираторных инфекций
При спинальной мышечной атрофии 2 типа также высок риск развития дыхательной недостаточности . Прогрессирование симптомов настолько разнообразно, что некоторые пациенты умирают в младенчестве, другие в состоянии достичь зрелости .
3 тип – болезнь Кугельберга-Веландера
Детская форма спинальной мышечной атрофии, которая:
- может возникнуть в возрасте от полутора лет
- по сравнению с предыдущими случаями, дети могут стоять и ходить самостоятельно, эта способность в некоторых случаях сохраняется до зрелого возраста
- наблюдается тремор рук и могут возникнуть проблемы с суставами и сколиоз
- нарушения дыхания и глотания проявляются менее часто, чем при 1 и 2 типе
У людей, страдающих от 3 типа спинальной мышечной атрофии, средняя продолжительность жизни сравнима со здоровыми людьми . Но, из-за проблем с питанием и низкой физической активность, часто имеют избыточный вес.
4 тип спинальной мышечной атрофии
Это «взрослая» форма спинальной мышечной атрофии, более мягкое и менее распространенное заболевание. Обычно возникает после 35 лет и медленно прогрессирует , затрагивая, в частности, способность передвигаться. Могут появиться судороги и проблемы с дыханием .
Продолжительность жизни нормальная.
Как распознать спинальную мышечную атрофию
Специалист по детской неврологии задаст ряд вопросов, чтобы получить подробный отчет о медицинской истории ребенка и его семьи, после процедуры физического обследования , чтобы оценить физическое состояние маленького пациента.
Подтверждение диагноза спинальной мышечной атрофии достигается благодаря генетическому тесту : берут образец крови и исследуют на наличие аномального гена SMN1. Тест можно использовать, чтобы найти носителей.
Поиск неисправного SMN1 также может осуществляться путём биопсии ворсинок хориона , которые являются частью плаценты, что делает возможным пренатальную диагностику в случае:
- если у пары уже был ребёнок, пострадавший от спинальной мышечной атрофии
- партнеры обнаруживают, что являются носителями, но все равно хотя родить ребёнка
Иногда бывает, что нельзя точно утверждать, что это спинальная мышечная атрофия. Тогда используют другие тесты , которые помогают провести дифференциальный диагноз между спинальной атрофией и другими патологиями нервов и мышц:
- электромиография , которая измеряет электрическую активность мышц
- мышечная биопсия , то есть изучение образцов мышечной ткани
- оценка концентрации креатина киназы , фермента уровень которого повышается при повреждении мышц
Как облегчить симптомы спинальной атрофии
На данный момент не существует лекарств для лечения спинальной мышечной атрофии, поэтому пациенты могут воспользоваться только поддерживающим лечением .
Поддерживающая терапия
Базируется на трёх «краеугольных камнях»:
- диетология
- дыхание
Для пациентов школьного возраста важно, чтобы они активно участвовали в школьных мероприятиях, потому что их физическая инвалидность никак не влияет на способности к обучению.
Физиотерапия
Физиотерапия необходима независимо от возраста человека. Упражнения позволят максимизировать амплитуду движений, чтобы предотвратить или замедлить потерю мелкой моторики. Дети со спинальной мышечной атрофией 1 и 2 типа получают огромную пользу от гимнастики в бассейне, поскольку вода помогает стимулировать всю мышечную массу.
Пациентам с 3 типом спинальной мышечной атрофии нужны ортопедические устройства (инвалидные коляски, параподы и т.д.), которые обеспечивают удобство и мобильность. Упражнения также важны, поскольку помогают предотвратить сколиоз, который усугубляет проблемы с дыханием и движениями.
Диетология
Каждый человек, страдающий от спинальной мышечной атрофии, должен иметь свой индивидуальный план питания, чтобы предотвратить последствия недостаточного или избыточного питания.
У тех детей, которые имеют большие трудности при грудном кормлении, пережевывании пищи и глотании, нужно принять меры, чтобы избежать таких осложнений, как аспирационная пневмония.
- Вы можете прибегнуть к использованию назогастрального зонда, который проходит через нос и доставляет пищу в желудок. Его относительно легко установить и снять, но он может протекать, тогда его следует заменить
- Другой вариант – гастростомия, то есть вывод трубки из желудка; является более простым в обслуживании, но процедура выполняется в операционной под наркозом.
Дыхание
Есть в этом случае для со спинальной мышечной атрофией существует три цели:
- пациенты и всех люди, которые вступают в контакт с ними, должны быть вакцинированы, например, против вируса гриппа , пневмококковой инфекции и бактерии коклюша , потому что инфекции дыхательных путей могут быть очень опасными для таких пациентов
- если кашлевый рефлекс слабым, это можно исправить с помощью специального устройства (Cough Assist): оно создает быстрое изменение давления снаружи и внутри легких, и быстрое прохождение воздуха по дыхательным путям, что имитирует кашель, освобождая дыхательные пути от секрета и микробов
- наконец, важна оценка дыхательной функции этих субъектов по степени сатурации кислорода в крови. Если количества кислорода меньше потребностей, стоит серьёзно рассмотреть идею использования механического респиратора. Изначально он используется в случае инфекции дыхательных путей и во время сна; с развитием атрфоии – весь день.
Возможные терапевтические стратегии
Открытие причины заболевания открыло для исследовательских групп большое направление для поиска методов лечения, направленных на максимально возможное замедление прогрессирования симптомов : повышение уровня белка SMN.
- Поскольку спинальная мышечная атрофия является моногенным заболеванием, это позволяет вмешаться в корень недуга, предоставляя пациентам функционирующий ген SMN1 (генная терапия)
- У лиц, страдающих от спинальной атрофии, но имеющих ген SMN2, можно увеличить экспрессию этого гена и заблокировать исключение экзона 7 во время сплайсинга незрелой мРНК.
В обоих случаях количество функционирующего белка SMN увеличивается .
AVXS-101 – экспериментальный препарат, разработанный биотехнологической компании Авексис, которому удалось достичь 1 этапа экспериментов на людях, при оценке безопасности лечения, и он начинает проверку эффективности.
Авексис сосредоточились на детях, страдающих от спинальной мышечной атрофии 1 типа, потому что это самый распространенный и смертельный тип заболевания.
AVXS-101 состоит из большого числа частиц адено-ассоциированного вируса серотипа 9, неспособного к репликации, но содержащего одну копию нормального гена SMN1.
Вводится в организм внутривенно, в состоянии преодолеть гематоэнцефалический барьер и достичь моторных нейронов.
Молекула ДНК, переносимая каждым вирусным вектором, производится в лаборатории. Она не изменяет ДНК пациента; содержит промотор, т.е. последовательность, которая способствует транскрипции ДНК в РНК, и гарантирует постоянное производство протеина SMN.
- AVXS-101 хорошо переносится маленькими пациентами с атрофией 1 типа
- ни один ребенок не испытал «события»: событие – это смерть или использование механического респиратора в течение 16 часов каждый день в течение 2 недель подряд не связанного с острой респираторной инфекцией
- было отмечено улучшение двигательных навыков
- 100% из пациентов, которые не имели сложностей с кормлением, оставались стабильными
- 8 из 10 детей, которые вошли в исследование без проблем с дыханием, по-прежнему не нуждаются в поддержке
Спинальной амиотрофией Верднига-Гоффмана называется наследственная патология нервов, которая поражает тот их участок, что контролирует скелетные мышцы. Ее характеризует слабость сразу всех мышц в организме. Более половины нервных клеток, которые контролируют мышцы, располагаются в спинном мозге. Именно потому заболевание названо «спинальным». «Мышечным» оно также названо из-за пагубного влияния на мышцы: те не получают от этих самых нервов никаких сигналов. «Атрофия» является медицинским термином, который обозначает истощение или усыхание того, что не используется. Здесь это касается недействующих мышц. Человек с подобной болезнью не имеет никакой возможности ни сидеть, ни передвигаться, ни даже обслуживать себя. Вылечить это невозможно. Проведя дородовую диагностику, можно предотвратить рождение малыша с таким заболеванием. Здесь мы поговорим, как эта патология наследуется, каковы ее проявления, а также о том, как можно помочь больному человеку.
Спинальная амиотрофия Верднига-Гоффмана названа в честь двоих ученых, первыми ее описавших. Во второй половине XIX столетия они доказали морфологическую сущность болезни. Сначала оба ученых считали, что у данной патологии только одна форма. Но уже в XX столетии ученые Кукельберг и Веландер открыли другую ее клиническую форму, с генетической причиной, схожей с той, которая и у открытой Верднигом и Гоффманом. Сейчас известно несколько клинических форм спинальной амиотрофии. Их объединяет общий наследственный дефект.
Эта патология наследственная. Она основана на мутации в пятой хромосоме. Мутирует тот ген, благодаря которому синтезируется белок SMN. Данный белок необходим для того, чтобы двигательные нейроны развивались именно так, как нужно. Стоит пятой хромосоме мутировать, и это негативно скажется на двигательных нейронах, помешав их развитию, а то и вовсе их разрушив. В результате мышца не может получать управляющие сигналы от нервов, а значит, не может и функционировать. Получается, не выполняется ни одно связанное с ней движение.

У мутировавшего гена аутосомно-рецессивный тип наследования. Фраза расшифровывается так: для развития спинальной амиотрофии нужно, чтобы у обоих родителей был мутантный ген. Если проще, то заболевание не разовьется, если хоть один из родителей не был носителем мутировавшего гена. В то же время они сами не болеют: у людей гены парные, а у отца и у матери ребенка доминирует здоровый ген. В таком случае больной малыш рождается примерно в четверти случаев. Ученые подсчитали, что около 2% живующих людей - носители гена с такой мутацией.
Классификация
Известны три разновидности данной патологии.
- Самая тяжелая, проявляющаяся раньше остальных.
- Среднетяжелая.
- Самая легкая, проявляющаяся в самом позднем возрасте.
По мнению некоторых врачей, существует еще одна разновидность: умеренная/мягкая СМА, которая проявляется уже у взрослого человека.
Стоит отметить, что кроме спинальной амиотрофии Верднига-Гоффмана существуют и другие типы СМА, которые отличаются симптомами и типами наследования. Они указаны в таблице ниже.
Таблица №1. Виды СМА.
| Название CMA | Тип наследования | Особенности и симптомы |
|---|---|---|
| SMAX1 | Х -сцепленный рецессивный | Наблюдается в основном y пожилых, поражает бульбарные нервы чepeпa, вызывает нисходящий паралич. |
| SMAХ2 | Х - сцепл. рецессивный | Врождённая агрессивная форма, приводящая к смерти до 3 - х мес. Вызывает слабость, арефлексию, контрактуры и переломы. |
| SMAX3 | Х - сцепл. рецессивный | Поражает в основном мальчиков. Атрофия всех дистальных мышц. Медленное нарастание симптомов. |
| Дистальная ДCMA1 | Аутосомно - рецессивный | Врождённая, поражаются в основном руки, возможны тяжелее дыхательные нарушения. |
| Дистальные формы ДCMA2 - ДCMA5 | Аутосомно - рецессивный | Все четыре формы отличаются медленным прогрессированием, ДCMA5 диагностируется y молодых. |
| Ювенильная SMA (тип HMN1) | Аутосомно-доминантный | Встречается в юности |
| Врождённая спинальная aмиoтpoфия | Аутосомно-доминантный | Нарушение иннервации и атрофия бедер, стоп, коленей c контрактурой и деформацией; иногда поражается голосовые связки. |
| SMA Финкeля | Аутосомно-доминантный | Начинается преимущественно в 35 - 37 лет, но зафиксированы случаи заболевания и в детском возрасте. Медленно развивается вначале в ногах, a затем в руках. Активность и рефлексы снижены, наблюдается непроизвольное дрожание (фасцикуляция). |
| SMA (тип LED1) | Аутосомно-доминантный | Атрофия нижних конечностей y новорождённых. |
| CMA c вpoждeнными кocтными пepeлoмaми | Аyтocoмнo-peцeccивный | Тяжелее симптомы, как про бoлeзни. Вepднигa-Гoффмaнa, отягощённые переломами. |
| CMA c гипоплазией | Аутосомно-доминантный | Врождённая аномалия головного мозга c церебральными симптомами, микроцефалией и задержкой развития. |
Симптомы патологии
Врожденная разновидность заболевания (СМА I) начинает проявляться до того, как малышу исполнится полгода. До этого такой ребенок может вяло двигаться. Не так уж редко спинальную амиотрофию у малыша можно заметить еще в самом начале постэмбрионального периода его жизни - у него угасают глубокие рефлексы:
- у малыша недостаточно громкий крик;
- ему трудно сосать;
- он не может держать головку.

Определить такую патологию у младенца можно с первых дней жизни
Если подобное становится заметно позже, что происходит редко, малыш может научиться держать головку, а то и сидеть, однако патология быстро сводит такие умения к нулю. Характерно и следующее:
- ранние проблемы с речью;
- ухудшение глоточного рефлекса;
- фасцикулярные подергивания языка.
Этот вид патологии может прилагаться к олигофрении, а также к патологиям развития скелета:
- деформациям грудной клетки (воронкообразной/килевидной ее форме);
- искривлению позвоночника (сколиозу);
- суставным контрактурам.
Нередки и другие врожденные заболевания. Например:
- гемангиомы;
- гидроцефалия;
- косолапость;
- дисплазия тазобедренных суставов;
- крипторхизм.
СМА I самая «вредная»: ее сопровождают развивающийся паралич, а также парез мускулатуры, отвечающей за дыхание. Из-за последнего развивается дыхательная недостаточность, из-за которой больной даже может погибнуть. А нарушение глотания может оказаться причиной попадания пищи в дыхательные пути и развития аспирационной пневмонии. Она тоже может привести к смерти.
СМА II начинает проявляться после того, как малышу исполняется полгода. В таком возрасте малыш уже удовлетворительно развит, он может стоять, держать головку, садиться, переворачиваться. Однако почти никогда больной малыш не успевает овладеть навыками ходьбы. В большей части случает данное заболевание проявляется после того, как малыш перенесет какую-либо острую инфекционную патологию. Например, пищевую токсикоинфекцию.
Когда СМА II только начинает проявляться, у малыша в ножках появляются периферические парезы, которые потом быстро оказываются в ручках, в туловище. Проявляется диффузная гипотония мышц, начинают исчезать глубокие рефлексы. Можно заметить и следующее:
- контрактуры сухожилий;
- тремор пальцев;
- фасцикуляции (непроизвольное дерганье) языка.
Важно! Позже начинает проявляться бульбарный синдром, развивается дыхательная недостаточность. Благодаря замедленному, если сравнивать с врожденной формой, развитию этой разновидности СМА, больной человек обычно доживает до пятнадцати лет.
СМА III , она же амиотрофия Кугельберга-Веландера - наименее вредная разновидность спинальной амиотрофии. Начинает проявляться, когда ребенку исполняется два года. Иногда может протекать бессимптомно даже до 30 лет. Впрочем, в этом случае пациент уже далеко не ребенок, что, однако, не делает его здоровее. При СМА III не задерживается развитие психики, пациент долго может двигаться без посторонней помощи. У него даже есть шансы и в глубокой старости иметь возможность самостоятельно себя обслуживать.
СМА IV , она же взрослая форма спинальной амиотрофии, представляет собой медленно развивающуюся патологию. Она обычно начинается после того, как человеку исполняется 35 лет. Такая форма если и сокращает жизнь, то незначительно. С другой стороны, у больного наблюдаются следующая картина:
- слабые проксимальные мышцы;
- фасцикуляции;
- ухудшение сухожильных рефлексов;
- потеря способности к ходьбе.

Электромиограмма показывает «ритм частокола» - спонтанную ритмическую активность. Так можно выявить патологию передних спинномозговых рогов. Если провести морфологическое исследование биоптатов мышц, можно заметить атро- и гипертрофированные волокна первого и второго типов. Также скапливаются мелкие круглые волокна, которые перемежаются с волокнами гипертрофированными - это «пучковая» атрофия. Если провести патоморфологическое исследование, становится заметно набухание/сморщивание/атрофия мотонейронов спинномозговых передних рогов, а довольно часто - еще и ядер нервов, которые выходят из головного мозга.
Как диагностируют данную патологию?
Чтобы поставить правильный диагноз, невролог должен иметь информацию о том, в каком возрасте у человека появились первые симптомы, как быстро они прогрессируют. Также он должен выяснить неврологические данные. Причем здесь важнее всего информация о том, нет ли у пациента периферических двигательных нарушений вместе с полным сохранением чувствительности. Еще неврологу важно узнать, не имеет ли больной врожденных отклонений, костной деформации. Амиотрофию Верднига-Гоффмана может диагностировать и неонатолог. При помощи дифференциальной методики можно диагностировать:
- миопатию;
- развивающаяся мышечная дистрофия Дюшенна;
- боковой амиотрофический склероз;
- сирингомиелия;
- полиомиелит;
- синдром вялого ребенка;
- обменные патологии.
Чтобы подтвердить результаты диагностики, делают электронейромиографию. Так называется обследование нервно-мышечного аппарата. Оно позволяет обнаружить изменения, нужные, чтобы исключить первично-мышечный вид заболевания, а также убедиться в наличии болезни мотонейрона. Биохимического анализа крови слишком мало, чтобы выявить сколь-либо серьезное повышение уровня креатинфоскиназы, которое бывает во время развития дистрофии мышц. Сделав МРТ/КТ позвоночника, изредка можно обнаружить атрофию передних рогов спинного мозга, однако при помощи этой методики можно убедиться в том, что другого спинального заболевания нет.

Окончательно ставится диагноз «амиотрофия Верднига-Гоффмана» только когда будут получены данные биопсии мышц, а также анализа ДНК. При помощи морфологического обследования мышечного биоптата можно выявить патогномоничную пучковую атрофию мышц, перемежающуюся зонами атрофии миофибрилл и здоровой ткани, отдельными гипертрофированными миофибриллами, а также местами соединительных разрастаний. В обязательные генетические исследования входит прямая и косвенная диагностика. Прямая методика позволяет выявить гетерозиготное носительство генной аберрации, а это необходимо при генетическом консультировании родственников пациентов, а также супругов, планирующих рождение ребенка. Во всем этом важно количественное исследование генов локуса СМА.
Проведя дородовую генетическую экспертизу, можно понизить риск появления на свет малыша, болеющего амиотрофией Верднига-Гоффмана. Чтобы получить генетический материал плода, следует применять инвазивные методики диагностики плода:
- амниоцентез;
- биопсию хориона;
- кордоцентез.

Обнаружив у плода амиотрофию Верднига-Гоффмана, следует поставить вопрос об аборте.
Дифференциальная методика диагностирования заболевания
По симптомам данную патологию можно перепутать с врожденной миопатией. Это плохой мышечный тонус. Для исключения мышечной гипотонии используется биопсия. На спинальную амиотрофию Верднига-Гоффмана похож острый полиомиелит. У этой патологии бурное, сопровождающееся резко повышенной температурой тела и множественными асимметричными параличами, начало. Через несколько суток острая фаза полиомиелита переходит в восстановительную. При гликогенозах, а также врожденных миопатиях тоже понижается тонус мышц.

Но эти патологии отличаются от спинальной амиотрофии тем, что их провоцируют не мутации гена, а следующие факторы:
- проблемы с метаболизмом;
- карцинома;
- гормональное нарушение.
При этом необходимо исключить и такое:
- болезнь Гоше;
- синдром Дауна;
- ботулизм.
Каким образом лечат данную патологию, а также как спрогнозировать ее дальнейшее протекание
Сегодня еще не существует этиопатогенетического лечения спинальной амиотрофии. Сейчас ее лечат, улучшая обмен веществ в периферической нервной системе, а также в мышцах, чтобы просто задержать развитие симптомов.
При этом в разных сочетаниях используются средства следующих групп:
- нейрометаболиты - лекарства, производимые из гидролизата свиных мозгов, гамма-аминомасляная кислота, а также пирацетам;
- средства, облегчающие передачу импульсов к мышцам - галантамин, сангвинарин, неостигмин, ипидакрин;
- медикаменты, улучшающие трофику миофибрилл - коэнзим Q10, L-карнитин, Церебролизин, Цитофлавин, Глутаминовая кислота, АТФ, Карнитина хлорид, Метионин, Калия оротат, Токоферола ацетат;
- витамины В - Мильгамма, Нейровитан, Комбилипен;
- анаболики - Ретаболил, Неробол;
- лекарства для улучшения проводимости импульсов между нервами и мышцами - Прозерин, Нейромидин, Дибазол, Галантамин;
- медикаменты для того, чтобы улучшить кровообращение - никотиновая кислота, скополамин.

Кроме того, при спинальной амиотрофии полезно делать ЛФК, а также проводить сеансы мягкого массажа.
Сегодняшние технологии являются большим подспорьем для облегчения жизни больных, а также их близких. В этом им помогают портативные аппараты ИВЛ, а также автоматизированные инвалидные коляски. Для улучшения подвижности больных существуют разнообразные методики ортопедической коррекции. А вот главные перспективы лечения СМА заключаются в непрерывно развивающейся генетике, а также в поиске возможностей использования генной инженерии для корректирования генетических заболеваний.
Анестезия
Если человеку вне зависимости от его возраста требуется операция, необходимо следовать определенным мерам предосторожности. Вся хирургическая бригада обязана быть в курсе того, что представляет собой СМА. Больше всего это касается анестезиолога.
В начале развития данной патологии в клетках мышц, не получающих нервных сигналов, порой возникают аномалии, обусловленные попытками мышц «дотянуться» до связанных с ними нервов. Из-за таких аномалий миорелаксанты, нередко используемые при хирургическом вмешательстве, могут оказаться опасны. Имея представление о подобных проблемах, можно подобрать безопасные препараты.
Диета при спинальной амиотрофии
На данный момент еще не подтверждено, что какая-либо диета приносит пользу при СМА.
По мнению большого количества родителей, диета, включающая в себя много белка или специальные добавки к пище, могут повысить силу мышц ребенка. Но, несмотря на очевидную необходимость хорошего питания для больного ребенка, еще не доказано, что ему нужен именно определенный рацион. Причем некоторые продукты даже могут нанести вред его организму.
К примеру, аминокислотное меню порой чревато еще большими проблемами у тех детей, у которых в организме слишком мало мышечной ткани. По мнению некоторых специалистов, при недостатке мышечной ткани, та не может правильно перерабатывать аминокислоты и тогда их уровень в крови повышается слишком сильно.

Некоторым детям полезнее питаться понемногу, причем чаще трех-четырех раз в день. Нужно просто разделить для больного все количество пищи, принимаемое здоровым сверстником больного за день, на несколько частей.
Среди больных СМА есть обладатели лишнего веса. Возможно, что причина этого кроется в слишком калорийном рационе вкупе с недостатком движения. Если есть возможность, больной должен, заручившись помощью врача и диетолога, привести свой вес в норму. Это важно не только для здоровья и внешности, но и для тех, кто ухаживает за таким больным. Ведь они каждый день помогают больному подниматься и перемещаться.
Важно! Есть врачи, советующие принимать отпускаемые без рецептов пищевые добавки. Например, креатин, коэнзим Q10. Сейчас ведутся исследования того, как креатин действует при данной патологии.
ЛФК
По мнению большинства врачей, комфортная физическая активность, если только не впадать в крайности, весьма полезна для самочувствия и здоровья больного СМА.
Суставам необходимо обеспечивать подвижность, а не подвергать их риску повреждений. При этом следует сохранять объем движений, чтобы сохранять суставы эластичными. При этом нужно поддерживать циркуляцию крови. Также, что важнее всего для детей, надо поддерживать мобильность высокой, чтобы исследовать окружающее.
Важно! Лучше всего проводить занятия в бассейне, наполненном водой, имеющей температуру 30-32 градуса тепла. Но, во-первых, человек, больной СМА, не должен заниматься плаванием сам, а во-вторых, нужно соблюдать определенные меры безопасности.
По мнению некоторых врачей, уделять массу внимания постепенному понижению в организме и без того недостаточного числа мотонейронов не так уж и нужно. Для определения, следует ли это учитывать, разрабатывая комплекс ЛФК, нужны исследования. Мнения профессионалов расходятся: одни считают, что перегрузить организм упражнениями невозможно, другие же — что, делая гимнастический комплекс «до посинения», можно форсировать отмирание оставшихся мотонейронов. Так что при ЛФК требуется аккуратность и гимнастику необходимо прекратить, не доводя себя до изнеможения.
Программы трудо- и физиотерапии полезны для людей любого возраста, если те хотят научиться максимально рационально использовать оставшиеся мышечные функции, а также разобраться, как лучше всего справляться с повседневными задачами.
Сейчас можно найти приспособления, которые полезны даже малышам – для изучения окружающего пространства. Маленьким детям может помочь все, что только придумал для решения этой задачи человеческий гений, начиная от ходунков и заканчивая ортезами.
Более того, есть семьи, которые самостоятельно придумывают и изготавливают свои приспособления, снабженные особыми функциями. К примеру, такие, у которых можно менять высоту, чтобы ребенок мог хоть ползать на полу, хоть сидеть на столе.
Человеку любого возраста, если у него СМА, ощутимо полезны приспособления для решения повседневных задач, не вызывающих затруднений у здоровых людей.
Способ предупреждения данной патологии
Для предупреждения амиотрофии Верднига-Гоффмана у новорожденного родители должны вовремя пройти диагностику генетических отклонений, а также, вынашивая ребенка, будущей матери следует провести еще и претональную ДНК-диагностику малыша. Если у того выявляется амиотрофия Верднига-Гоффмана - встает вопрос об аборте.

Как спрогнозировать развитие патологии
Сейчас спинальная амиотрофия Верднига-Гоффмана неизлечима. Прогнозы абсолютно неблагоприятны. Если такая проявляется у малыша в первые дни после его рождения, тот обычно умирает до того, как ему исполнится полгода. Если же симптомы начинают проявляться после трехмесячного возраста, средний возраст такого ребенка равняется паре лет. Максимальный возраст, до которого может дожить такой больной, равен восьми годам. Ранний детский тип этой патологии прогрессирует куда медленнее, и такой ребенок доживает до подросткового возраста - четырнадцати-пятнадцати лет.
И все же нынешние технологии могут позволить больным детям жить намного дольше. Благодаря вентиляции легких при помощи современных портативных аппаратов, а также питанию с помощью зонда, подающего пищу прямо в желудок, маленький человек может прожить еще долгие годы.
Видео — Генетические основы спинальной мышечной атрофии
Другие болезни - клиники в Москве
Выбирайте среди лучших клиник по отзывам и лучшей цене и записывайтесь на приём
Другие болезни - специалисты в Москве
Выбирайте среди лучших специалистов по отзывам и лучшей цене и записывайтесь на приём

